Machine Learning for Molecular Simulations
Machine learning (ML) has emerged as a transformative tool in computational chemistry and molecular simulations. From creating large-scale datasets that democratize quantum chemistry to developing algorithms that enable efficient sampling of rare events, ML is revolutionizing how molecular discovery is approached and complex chemical processes are understood. Dr. Hasyim’s work spans both ends of this spectrum: building foundational datasets and developing novel algorithms for challenging computational problems.
Open Molecules 2025: Large-Scale Dataset for ML-Driven Molecular Chemistry
A fundamental challenge in developing machine learning models for molecular chemistry is the lack of comprehensive, high-accuracy training data that spans the vast diversity of chemical space. Despite substantial efforts in data generation, existing molecular datasets have been limited in either their scale, chemical diversity, or level of theoretical accuracy, making it difficult to train ML models that perform reliably across the broad range of molecular systems encountered in real-world applications.
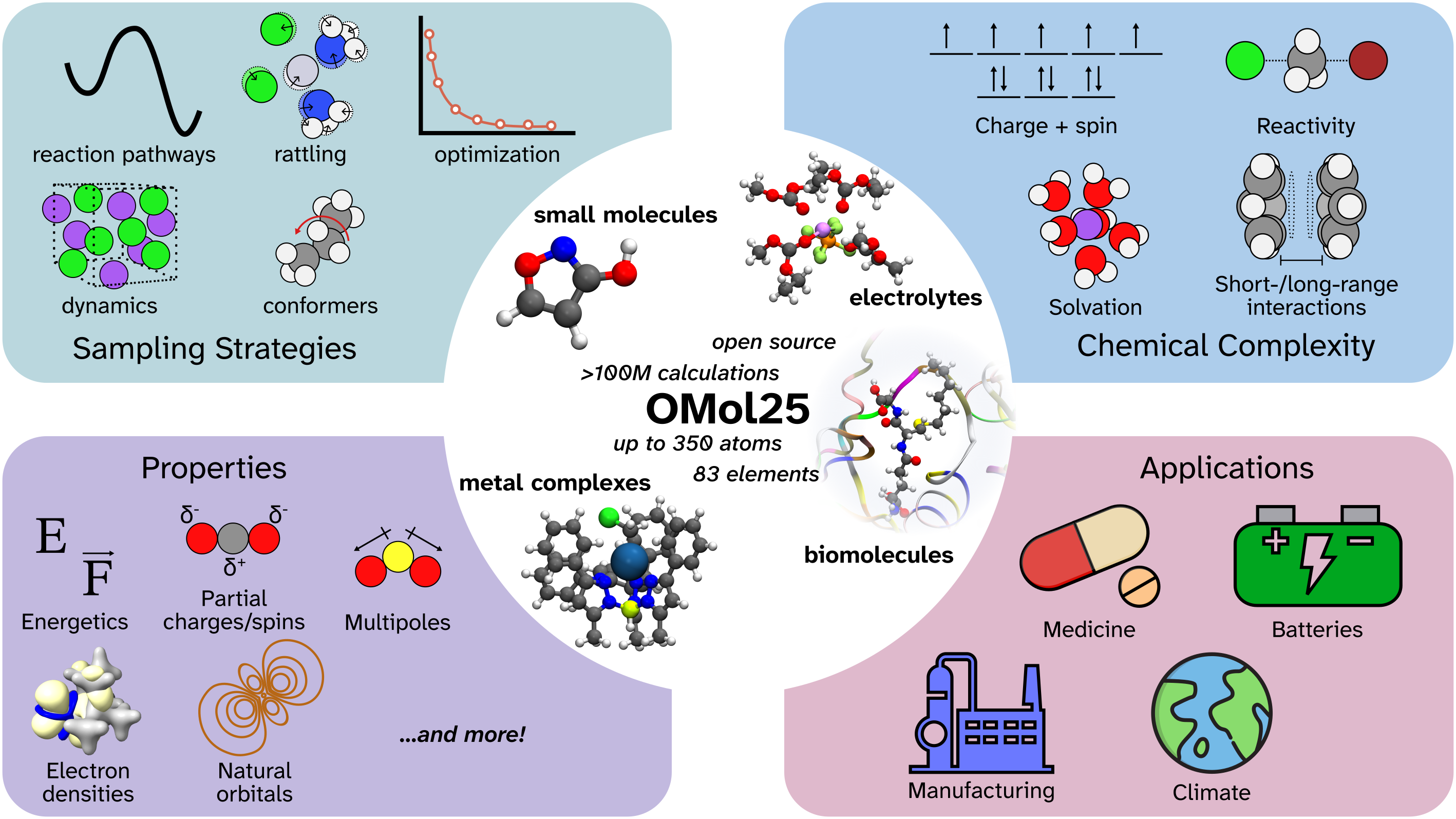
To address this critical gap, Dr. Hasyim collaborated with Meta FAIR and a broad consortium of researchers to develop Open Molecules 2025 (OMol25), a large-scale dataset composed of more than 100 million density functional theory (DFT) calculations at the ωB97M-V/def2-TZVPD level of theory. This dataset represents billions of CPU core-hours of compute and encompasses an unprecedented breadth of molecular chemistry, including 83 elements across the periodic table and systems containing up to 350 atoms.
What makes OMol25 unique is its comprehensive coverage of chemical diversity. The dataset includes approximately 83 million unique molecular systems spanning small molecules, biomolecules, metal complexes, and electrolytes. Beyond just structural diversity, OMol25 captures the full range of molecular complexity relevant to real-world applications: variable charge states and spin multiplicities, explicit solvation effects, conformational diversity, and reactive structures. This systematic approach to data curation ensures that ML models trained on OMol25 can generalize across the diverse landscape of molecular chemistry.
The dataset also pushes beyond the size limitations of typical quantum chemistry datasets, incorporating molecular systems that are substantially larger than those found in existing resources. This enables the development of ML models capable of handling the complex, multi-component systems encountered in drug discovery, materials science, and catalysis applications.
This collaborative effort with Meta FAIR represents a significant step toward democratizing high-accuracy molecular simulations and enabling the next generation of ML-driven molecular discovery. The public release of OMol25, along with baseline models and comprehensive evaluation benchmarks, provides the community with the resources needed to develop more accurate and generalizable ML models for molecular chemistry.
This work can be found in Ref. [1]. The collaboration represents a large-scale effort with Meta FAIR and researchers from across the computational chemistry and machine learning communities.
Computing Committor Functions with Neural Networks
The discovery of transition pathways governing kinetic processes at the microscopic level is of fundamental interest in chemistry. Common to these problems is the presence of high-energy barriers, which make the pathway discovery difficult due to the need to sample rare events.
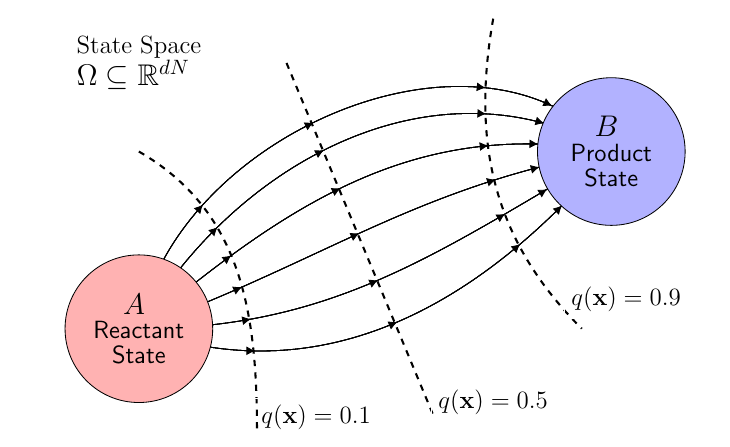
The committor function \(q(\mathbf{x})\), the probability that a trajectory starting from some initial configuration enters the product state before the reactant state, encodes the complete mechanistic details of these pathways, including the reaction rates and transition-state ensembles. Computing the committor function by traditional means is prohibitively expensive, requiring hundreds of trajectories per configuration to yield sufficient estimates along with the need for rare configurations along the transition pathway.
Dr. Hasyim’s work [2] extends the previous approach of Grant Rotskoff and Eric Vanden-Eijnden that combines importance sampling, in which high probability regions of the transition pathway are targeted, and machine learning, in which an artificial neural network approximates the committor function.
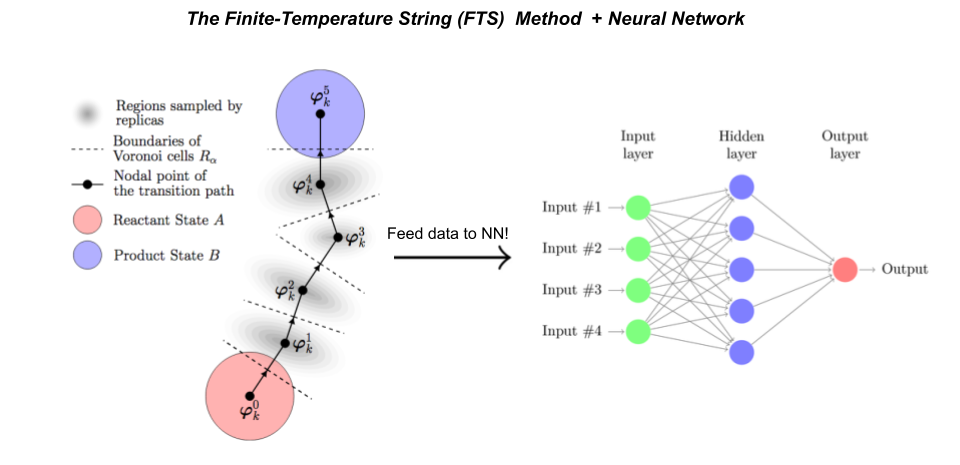
Dr. Hasyim and collaborators improved their method by adding two elements: (1) supervised learning, in which sample-mean estimates of the committor function obtained via short simulation trajectories are used to fit the neural network, and (2) the finite-temperature string method, a path-finding algorithm that enables homogeneous sampling across the transition pathway.
The research team then demonstrated these modifications on low-dimensional systems first, showing that they yield accurate estimates of the committor function and reaction rates. Additionally, an error analysis for their algorithm was developed from which the reaction rates can be accurately estimated via a small number of samples.
This work is done in collaboration with Clay H. Batton and under the supervision of Prof. Kranthi Mandadapu. Code may be found on Dr. Hasyim’s Github. Paper can be found in Ref. [2]